Acteurs de la Pathogénèse des Infections Rétrovirales
 L’équipe en Janvier 2021 De gauche à droite : N Chazal, V Hebmann, JM Mesnard, C Mourouvin, L Espert, M Jansen, JM Peloponese, MA Houmey, J Tram, M Abrantes, B Beaumelle, B Pradel, N Audemard, C Silvestre, L Marty, A Gross.
L’équipe en Janvier 2021 De gauche à droite : N Chazal, V Hebmann, JM Mesnard, C Mourouvin, L Espert, M Jansen, JM Peloponese, MA Houmey, J Tram, M Abrantes, B Beaumelle, B Pradel, N Audemard, C Silvestre, L Marty, A Gross.
Projets
Groupe 1 : Rôles et Fonctions de la protéine HBZ dans l’établissement de l’ATL (Responsable Jean-Marie Peloponese) Groupe 2 : ASP, la protéine anti-sens du VIH-1 : évolution, impacts immunologique, cellulaire et viral (Responsable : Antoine Gross) Groupe 3 : Rôle de la protéine Tat extracellulaire lors du SIDA – (Responsable : Bruno Beaumelle)
Groupe 1 : Rôles et Fonctions de la protéine HBZ dans l’établissement de l’ATL
Le virus T-lymphotropique humain (HTLV-1) a été le premier rétrovirus trouvé pour induire des maladies chez l’homme. HTLV-1 provoque une maladie néoplasique agressif, la leucémie des cellules T adultes (ATL), les maladies neurodégénératives, telle que la myélopathie associé à HTLV-1 / ou paraparésie spastique tropicale (TSP / HAM) et les maladies inflammatoires telles que l’uvéite. HTLV-1 infecte environ 15 millions de personnes dans le monde. HTLV-1 est endémique des régions intertropicales comme le Brésil, le bassin caribéen, l’Afrique sub-saharienne et le sud du Japon. HTLV-1 est un rétrovirus complexe, qui code pour des gènes de régulation (Tax, Rex et HTLV-1 facteur de bZIP (HBZ)) et plusieurs gènes auxiliaires, tels que p30, p12, p13. Parmi ceux-ci, Tax a été proposé pour jouer un rôle central dans la transformation des cellules infectées. Cependant, étant donné qu’il est une cible majeure des lymphocytes T cytotoxiques, son expression est souvent réduite au silence dans les cellules d’ATL afin d’échapper au système immunitaire de l’hôte. Le gène hbz, caractérisé dans notre laboratoire en 2002, est codé par le brin moins du génome viral. Curieusement, alors que Tax est réprimé dans les cellules infectées, l’ARNm d’HBZ a été détecté dans toutes les cellules d’ATL. HBZ est une petite protéine nucléolaire qui contient un domaine d’activation en N-terminal et un « Fos-like Leucine Zipper » en C-terminal. Nous avons montré que HBZ est en mesure de maintenir sa propre expression dans les cellules infectées via une boucle de régulation comprenant JunD et SP1. Nous avons également montré que HBZ d’induire la prolifération cellulaire et de transformer des cellules in vitro. Nos thématiques de recherche sont principalement centrées sur l’étude des mécanismes cellulaires et moléculaires par lesquels HBZ transforme la cellule infectée. Nous nous concentrons actuellement sur 2 axes : – nous étudions l’importance des interactions entre HBZ et la voie AP-1 dans les cellules infectées par HTLV-1. – en collaboration avec des cliniciens, nous essayons de comprendre le rôle de HBZ dans la chimiorésistance des cellules d’ATL et de développer de nouvelles approches thérapeutiques contre HBZ.
Groupe 2 : ASP, la protéine anti-sens du VIH-1 : évolution, impacts immunologique, cellulaire et viral
Cet axe de recherche porte sur une protéine méconnue du VIH-1, ASP (AntiSense Protein). Grâce à l’utilisation de plusieurs approches expérimentales (virologie, biologie cellulaire, immunologie, bio-informatiques, études chez les patients infectés par le VIH-1), notre projet vise à déterminer la fonction de cette protéine et à comprendre son rôle dans la pathogenèse du SIDA (latence virale, chronicité). Le VIH-1 produit l’ensemble de ses protéines à partir de l’ADN proviral intégré (constitué notamment de deux LTR5’ et un LTR3’) grâce à une activité transcriptionnelle d’un promoteur localisé dans le LTR-5’ (Long Terminal Repeat). A l’instar du HTLV-1 qui possède une activité transcriptionnelle conduisant à la production de la protéine HBZ (cf. projet HBZ), nous avons montré que le LTR-3’ du VIH-1 possédait également une activité transcriptionnelle anti-sens permettant la production d’une protéine, la protéine ASP. 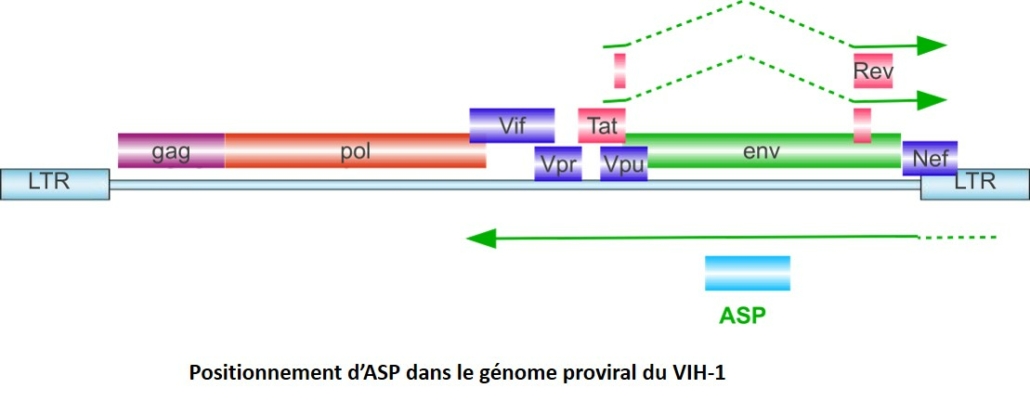 Bien que son existence ait été proposée pour la première fois en 1988, avec l’observation d’un cadre ouvert de lecture sur le brin antisens du génome proviral, en recouvrement du gène de l’enveloppe (Savoret et al., Frontiers in Microbiol, 2021), la fonction de la protéine ASP n’est pas à ce jour encore connue. Au cours des dernières années, nous avons pu caractériser le transcrit asp (Landry et al., Retrovirology, 2007), identifier la population cellulaire dans laquelle cette protéine s’exprime (Laverdure et al, J. Virol., 2012). Parallèlement à ces travaux, nous avons également pu montrer qu’ASP est exprimée chez les patients infectés, ceux-ci développant une réponse immunitaire anti-ASP cellulaire (Bet et al., Retrovirology, 2015) et humorale (Savoret et al., Frontiers in Microbiol., 2020). Enfin, grâce à une étude évolutive développée en collaboration avec des informaticiens du LIRMM et réalisée sur plus de 23 000 séquences, nous avons démontré que le gène asp était apparu sous sa forme actuelle lors de l’émergence de la pandémie de VIH-1 au début du vingtième siècle (Cassan et al., PNAS, 2016).
Bien que son existence ait été proposée pour la première fois en 1988, avec l’observation d’un cadre ouvert de lecture sur le brin antisens du génome proviral, en recouvrement du gène de l’enveloppe (Savoret et al., Frontiers in Microbiol, 2021), la fonction de la protéine ASP n’est pas à ce jour encore connue. Au cours des dernières années, nous avons pu caractériser le transcrit asp (Landry et al., Retrovirology, 2007), identifier la population cellulaire dans laquelle cette protéine s’exprime (Laverdure et al, J. Virol., 2012). Parallèlement à ces travaux, nous avons également pu montrer qu’ASP est exprimée chez les patients infectés, ceux-ci développant une réponse immunitaire anti-ASP cellulaire (Bet et al., Retrovirology, 2015) et humorale (Savoret et al., Frontiers in Microbiol., 2020). Enfin, grâce à une étude évolutive développée en collaboration avec des informaticiens du LIRMM et réalisée sur plus de 23 000 séquences, nous avons démontré que le gène asp était apparu sous sa forme actuelle lors de l’émergence de la pandémie de VIH-1 au début du vingtième siècle (Cassan et al., PNAS, 2016). 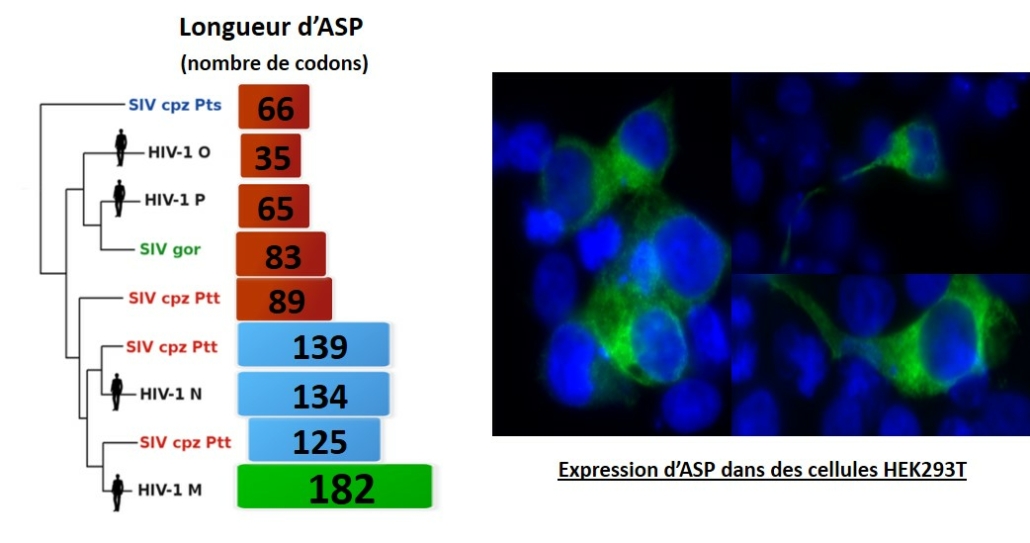 Nos recherches actuelles portent plus particulièrement sur : – L’étude de la fonction d’ASP et son lien avec l’infection et la réponse immune. – Le lien entre ASP et le statut immunitaire du patient infecté sous traitement anti-rétroviral. – Les mécanismes évolutifs d’ASP chez le VIH-1. COVID-19 : Plus récemment, nous avons initié des études sur le SARS-COV2, en particulier sur certaines protéines du virus peu étudiées en nous intéressant à des aspects de biologie cellulaire et à la réponse anticorps chez les patients. Contrats obtenus ces dernières années : – Fédération Hospitalo-Universitaires Infection Chronique (FHU Inch) : 2020-2023 : Contrat doctoral (3ans) en collaboration avec le Dr. Alain Mackinson du Service de maladies infectieuses et tropicales du CHU. – Sidaction : 2020-2023 : contrat doctoral (3 ans) – Sidaction : 2019 : financement jeune chercheur (1 an) : 4ième année de thèse – Direction des relations avec les entreprises du CNRS : 2019-2020 – UM/CHU : 2015-2018 : Contrat doctoral en santé (3 ans) – CNRS mission Interdisciplinarité 2013-2016
Nos recherches actuelles portent plus particulièrement sur : – L’étude de la fonction d’ASP et son lien avec l’infection et la réponse immune. – Le lien entre ASP et le statut immunitaire du patient infecté sous traitement anti-rétroviral. – Les mécanismes évolutifs d’ASP chez le VIH-1. COVID-19 : Plus récemment, nous avons initié des études sur le SARS-COV2, en particulier sur certaines protéines du virus peu étudiées en nous intéressant à des aspects de biologie cellulaire et à la réponse anticorps chez les patients. Contrats obtenus ces dernières années : – Fédération Hospitalo-Universitaires Infection Chronique (FHU Inch) : 2020-2023 : Contrat doctoral (3ans) en collaboration avec le Dr. Alain Mackinson du Service de maladies infectieuses et tropicales du CHU. – Sidaction : 2020-2023 : contrat doctoral (3 ans) – Sidaction : 2019 : financement jeune chercheur (1 an) : 4ième année de thèse – Direction des relations avec les entreprises du CNRS : 2019-2020 – UM/CHU : 2015-2018 : Contrat doctoral en santé (3 ans) – CNRS mission Interdisciplinarité 2013-2016
Groupe 3 : Rôle de la protéine Tat extracellulaire lors du SIDA
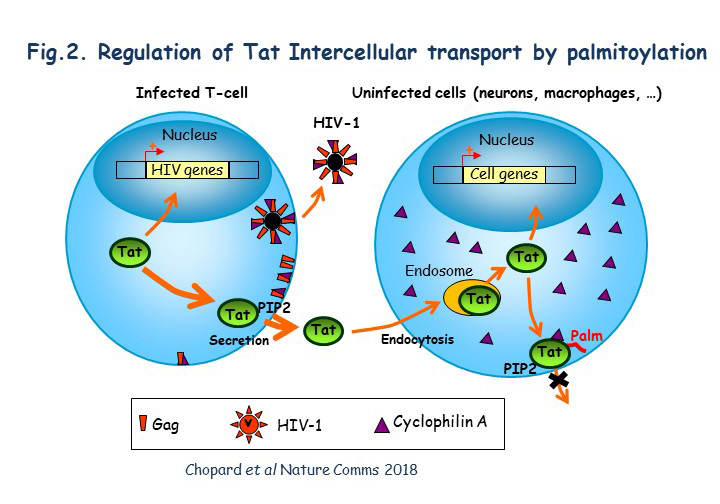
*Travaux antérieurs Notre groupe s’intéresse au transport intracellulaire et transcellulaire de la protéine Tat du VIH. Cette protéine est bien connue et étudiée car elle est indispensable à la transcription des gènes viraux. Son taux et son niveau d’activation régule la sortie de latence du VIH. Nous avons montré que Tat est sécrétée de manière non conventionnelle après fixation sur le PI(4,5)P2 sur le feuillet interne de la membrane plasmique (Rayne et al, 2010). La Tat circulante peut alors se fixer sur les cellules via un certain nombre de récepteurs dont le LRP (Liu et al, 2000). Nous avons montré que Tat est ensuite endocytée par la voie clathrine (Vendeville et al, 2004) et qu’une fois dans les endosomes le pH acide va déclencher un changement de conformation induisant l’insertion membranaire de Tat grâce à son résidu Trp unique (Trp11) (Yezid et al, 2009). Sa translocation vers le cytosol est catalysée par Hsp90, une chaperone cytosolique (Vendeville et al, 2004). Une fois dans le cytosol Tat peut induire un certain nombre de réponses cellulaires, sécrétions de cytokine inflammatoires par les monocytes par exemple (Rayne et al, 2004). La Tat « entrante » va elle aussi se fixer sur le PI(4,5)P2. La Tat recombinante a une affinité pour ce phosphoinositide bien plus forte que celle des protéines cellulaires car Tat insère la chaine latérale de son Trp dans la membrane lors de la fixation du PI(4,5)P2 (Debaisieux et al, 2012). Dans les cellules non infectées, Tat va de plus être palmitoylée ce qui va encore stabiliser son association membranaire et inhiber sa sécrétion. Cette palmitoylation requiert la prolylisomérase cyclophiline A (CypA). Tat n’est pas palmitoylée dans les cellules infectées car la CypA est encapsidée par le VIH-1 (~200 CypA/virion), ce qui vide la cellule de sa CypA et permet la forte sécrétion de Tat par les cellules infectées (Chopard et al, 2018) (Fig.2). Dans les cellules non infectées, la palmitoylation de Tat la rend donc résidente sur le PI(4,5)P2, ce qui va fortement perturber l’assemblage de différentes machineries utilisant ce phosphoinositide. Nous avons ainsi montré que la Tat circulante inhibait le recrutement de l’annexine 2 et cdc42 perturbant ainsi respectivement la neurosécrétion (collaboration avec le groupe de Nicolas Vitale, INCI, Strasbourg (Tryoen-Toth et al, 2013) et la phagocytose (Debaisieux et al, 2015) (Fig.1). Tat perturbe aussi le fonctionnement certains canaux ioniques chez les cardiomyocytes (collaboration avec le groupe de Gildas Loussouarn, INSERM, Nantes) (Es-Salah-Lamoureux et al, 2016). Il faut noter que ces différents dysfonctionnements sont observés chez les patients VIH. Nos travaux sont résumés dans les Figs. 1 et 2. Ils ont été financés par l’ANRS, Sidaction et la FRM et réalisés par Fabienne Rayne (PostDoc), Agnés Vendeville, Hocine Yezid, Solène Debaisieux, Christophe Chopard, Bao Viet Tong et Malvina Schatz (doctorants). 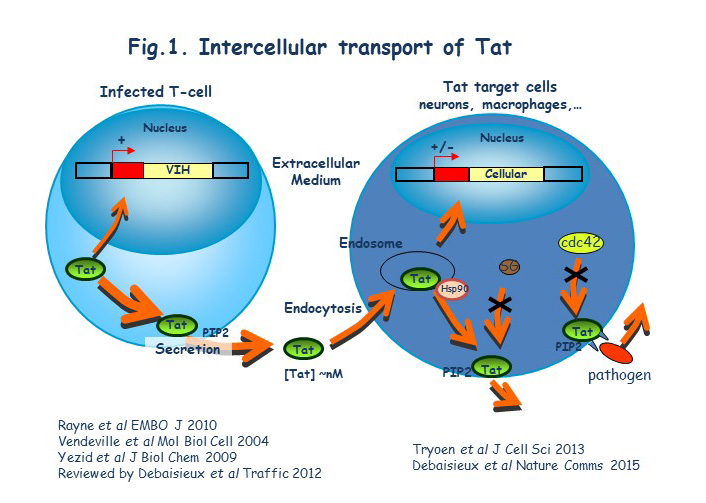
 *Travaux actuels Nous nous intéressons à l’effet de Tat sur la multiplication des pathogènes opportunistes lors du VIH (collaborations Laurent Kremer et Laura Picas de l’IRIM, Oliver Neyrolles et Christel Verollet de l’IPBS Toulouse), au mécanisme d’encapsidation potentiel de Tat (collaboration Mickaël Blaise et Laurent Chaloin de l’IRIM, Pierre-Emmanuel Milhiet et Jean-François Guichou, CBS Montpellier) et au développement de nouveaux agents de levée de latence du VIH (collaboration Laurent Chaloin), que nous venons de breveter. Ces travaux sont financés par Sidaction et la SATT AXLR.
*Travaux actuels Nous nous intéressons à l’effet de Tat sur la multiplication des pathogènes opportunistes lors du VIH (collaborations Laurent Kremer et Laura Picas de l’IRIM, Oliver Neyrolles et Christel Verollet de l’IPBS Toulouse), au mécanisme d’encapsidation potentiel de Tat (collaboration Mickaël Blaise et Laurent Chaloin de l’IRIM, Pierre-Emmanuel Milhiet et Jean-François Guichou, CBS Montpellier) et au développement de nouveaux agents de levée de latence du VIH (collaboration Laurent Chaloin), que nous venons de breveter. Ces travaux sont financés par Sidaction et la SATT AXLR.





En bref
L’équipe s’intéresse à la fois à des protéines virales dont le rôle est bien établi dans l’infection (Tat du VIH-1) ou la transformation cellulaire (Human bZIP factor de HTLV-1, HBZ), mais aussi à une protéine du VIH-1 dont le rôle est encore inconnu (la protéine antisens du VIH-1, ASP). Nos travaux visent à mieux cerner la fonction et l’activité biologique de ces protéines à la fois dans la multiplication virale et dans les effets pathogènes de l’infection. Nous étudions également la réponse cellulaire aux infections virales, notamment le processus d’autophagie.
Financements
![]()